Abstract
Background: Observational studies generate information on real-world therapy and complement data from prospective randomized trials. LEAN is an open-label, non-interventional, multi-centre, German cohort study on leuprorelin in routine clinical practice. Objectives: To extend knowledge on the use, effectiveness, and tolerability of HEXAL/Sandoz leuprorelin (in this article, the term Leuprone® HEXAL® covers Leuprorelin Sandoz® as well) solid implant in patients with prostate cancer (PCa) in a real-world setting. Methods: 959 PCa patients scheduled for androgen deprivation therapy (ADT) received leuprorelin acetate implant. Metabolism, serum prostate-specific antigen (PSA), and testosterone data, if available, were collected at baseline and follow-up visits for ≥12 months. Results: Of 694 patients in the modified full analysis set, 26.4% received GnRH analogues ≤6 months before enrolment. Fifty-one percent of patients were treated for locally advanced or metastatic PCa. In 19.6% of patients, ADT was used in neoadjuvant or adjuvant settings and in 28.5% with rising PSA after definite therapy. Testosterone levels <0.5 ng/mL were achieved in >90% of patients. Safety profile was in line with the summary of product characteristics. Therapy was well tolerated, with patient-triggered therapy discontinuation in 3.6%. Conclusions: This interim analysis confirmed previous efficacy findings for leuprorelin implant in a real-world setting. This contemporary cohort showed a shift in the use of ADT to non-metastatic PCa stages.
Introduction
For decades, androgen deprivation therapy (ADT) based on luteinizing hormone-releasing hormone (LHRH) agonists has represented a standard of care in men with advanced prostate cancer (PCa), suppressing blood testosterone to castrate levels in a vast majority of patients [1-3]. More recently, ADT has also been recommended in combination with radiotherapy in patients with intermediate- or high-risk localized/locally advanced PCa and in combination with docetaxel or abiraterone plus prednisone for men with metastatic disease. In addition, ADT may be used for biochemical tumour recurrence in high-risk patients after radical prostatectomy [3, 4].
Leuprorelin acetate, a synthetic analogue of LHRH, is the most widely prescribed drug of its class [1, 3, 5]. Two unique slow-release pharmaceutical forms of leuprorelin acetate (Leuprorelin Sandoz® and Leuprone® HEXAL®; 1-month and 3-month implant1) were first approved in 2007 [6].
Guideline recommendations are preferentially based on results obtained in prospective randomized trials; however, patients included in these trials represent a highly selected cohort. In conclusion, it needs to be questioned if these results can be translated to real-world patients being treated under much less controlled circumstances. Therefore, post-authorization observational studies must be considered as important sources of information on therapies that complement data obtained in phase III trials. Despite their design limitations, observational studies extend existing knowledge in particular on rare side effects and, if appropriately designed, may identify risk factors for unresponsiveness to therapy or address further important questions related to a given therapy not adequately covered in phase III trials.
The LEAN study (www.germanctr.de, trial ID: DRKS00005643, “Observation of Leuprone® HEXAL® in treatment practice. Non-interventional study on effectiveness and tolerability and on the influence of anamnestic factors”) is an open-label, multi-centre, non-interventional German cohort study of Leuprone HEXAL in routine clinical practice. This study was designed to extend existing knowledge on the efficacy and tolerability of leuprorelin solid implant in men with PCa and to explore the influence of patient and lifestyle factors on disease course [4, 7-9]. This manuscript also compares the real-world clinical use for LHRH agonists in a contemporary patient cohort with guideline-recommended indications.
Patients and Methods
Study Description
The primary objective of the study was to assess prostate-specific antigen (PSA) and testosterone concentrations in serum over time, investigate the current use of ADT, and describe the tolerability profile. Secondary objectives were to investigate associations between various patient factors and the course of PSA and testosterone, to analyze the real-world use of leuprorelin implant, and to capture the comorbidities and lifestyle of the patients. The HEXAL® leuprorelin solid implant is a depot formulation measuring approximately 10 × 1.5 mm that is administered subcutaneously.
Patient Selection Criteria
The study included patients with hormone-sensitive PCa, in whom treatment with Leuprone HEXAL (1 month or 3 months) was planned in accordance with the summary of product characteristics (SmPC). Further inclusion criteria were life expectancy >12 months and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2. There was no age limit. Patients were permitted to have received an LHRH agonist for <6 months before the start of the study; however, concomitant therapy with a gonadotropin-releasing hormone antagonist, abiraterone acetate, enzalutamide, or chemotherapy was not permitted. Local or palliative radiotherapy was allowed.
Patient Assessment and Data Collection
Patient characteristics, metabolic data (blood glucose, glycated haemoglobin, triglycerides, cholesterol, and albumin in urine), serum PSA, and testosterone (when available) were assessed at baseline. PSA and testosterone serum measurements were performed locally. Therefore, there is some variability in the automated immunoassays used at individual study sites. PSA and testosterone (if available) were also assessed during follow-up (planned for at least 12 months), and available measurements were included in the analysis.
Testosterone response was listed using cutoffs at 0.2 and 0.5 ng/mL (0.7 and 1.7 nmol/L) during ADT. For the overall analysis, each measurement (PSA or testosterone) was allocated to the nearest point in time of a monthly grid starting with the first dose of LHRH therapy. For the comparison with the PR-7 results, (1) all testosterone values were included and opposed to (2) those testosterone samples taken in the week before next Leuprone administration. Adverse events (AEs) were registered as reported by centres, and their suspected relation to Leuprone was documented verbatim and coded and analyzed using the Medical Dictionary for Regulatory Activities (MedDRA) classification.
Statistical Analysis
Patient characteristics, medical history, and demographic features were analyzed using descriptive statistics and presented as mean ± SD or frequency categories, unless stated otherwise. Cox-regression analyses were carried out to investigate the possible relationship between baseline factors and time to PSA progression and between baseline factors and time to tumour progression. A subgroup analysis was performed to evaluate the influence of age, body mass index, tumour staging, and other factors on PSA and testosterone values and tumour progression status. In addition, patients were categorized to subgroups depending on treatment intention and extent of disease (Table 1).
Results
Baseline Characteristics – Modified Full Analysis Set
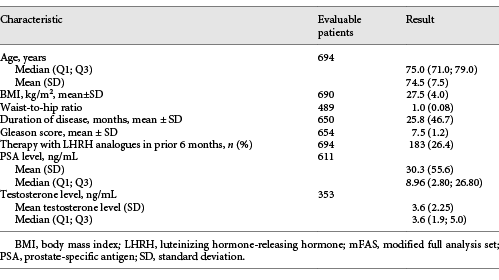
A total of 959 patients with PCa and a need for ADT, as decided by the urologist, were screened, and 921 patients were included in the trial at 150 centres from January 2014 to July 2015. For further analysis, all patients were considered after excluding patients who did not receive study treatment or had missing data, with 694 patients considered for the modified full analysis set (mFAS) (Fig. 1).
The median age was 75 years. On average, the primary diagnosis of PCa was made 25.8 months (SD: 46.7 months) before inclusion in the study. Demographic parameters are summarized in Table 1.
In a list of pre-defined disease categories, physicians indicated that 52% of patients suffered from concomitant cardiovascular diseases and 16% from endocrine/metabolic disorders. In an analysis of baseline lab values, a quarter or more of patients presented with borderline or elevated results for glucose and lipid metabolism. A third of patients had no documented relevant comorbidities at baseline.
Regarding ECOG status, 416/694 patients (59.9%) were considered ECOG 0 and another 233 patients (33.6%) were classified as ECOG 1. The remaining 45 patients (6.5%) received an ECOG 2 classification.
Indication
Based on the mFAS, the most frequent indications for ADT in the study cohort were a Gleason score of ≥6 at initial diagnosis, advanced tumour according to the Tumour, Node, Metastasis classification, and a most recent PSA of ≥10 ng/mL. Details are displayed in Table 2.
Treatment Efficacy
Serum PSA
The median PSA decreased from 9 ng/mL (mFAS) at baseline to 0.62, 0.31, 0.22, and 0.20 ng/mL at 3, 6, 9, and 12 months, respectively, after the start of ADT (Fig. 2). In subgroup analyses, the median PSA dropped from 7.96 to 0.11 ng/mL at 12 months in locally advanced and/or metastatic PCa, from 2.72 mg/mL to <0.1 ng/mL in patients after RP, and from 7.44 to <0.1 ng/mL in patients receiving ADT related to radiation therapy (neoadjuvant, biochemical tumour recurrence).
Serum Testosterone
Median serum testosterone levels decreased from 3.6 ng/mL at baseline to <0.2 ng/mL from month 1 onwards (Fig. 3). At baseline, the proportion of patients with testosterone ≤0.2 ng/mL was 5.9% and with ≤0.5 ng/mL was 11.3%. More than 90% of patients achieved testosterone levels ≤0.5 ng/mL at 3, 6, and 12 months after baseline. A testosterone breakthrough (testosterone serum levels of ≥0.5 ng/mL) was detected in 8.6, 4.1, 1.4, and 5.3% of measurements at 3, 6, 9, and 12 months. A separate exploratory analysis studying testosterone serum levels in relation to insertions found that testosterone surges were more frequent in the week before insertion of the next implant compared with 15–75 days after an insertion (7.6 vs. 4.3%).
Patient Safety
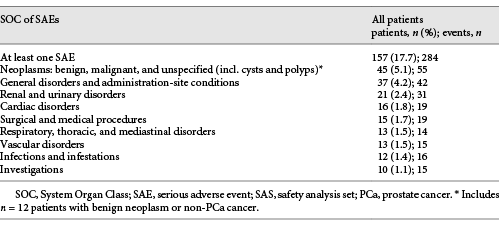
In summary, 270 patients (30.6%) reported 532 AEs and 157 patients (17.7%) experienced 284 serious AEs (SAEs; Table 3). A total of 104 adverse drug reactions (ADRs) were documented in 63 patients (7.1%), and 17 serious ADRs were documented in 10 patients (1.1%). Most frequent ADRs were hot flushes, hyperhidrosis, drug ineffective/drug effect decreased, and weight increase.
For most side effects, reported frequencies were either within the range suggested by the SmPC or somewhat lower. Dizziness and vertigo were reported in 2 (0.2%) and 3 (0.3%) of all 886 patients in the safety analysis set (SAS), hypertension in 0.2%, and pulmonary embolism in 0.5%. This is slightly higher than the labelled frequency of 0.01–0.1%. Because of the low number of such AEs, this evidence is currently inconclusive.
The great majority of patients (86.6%, n = 601/694) completed the study as planned in the study protocol. Premature termination took place at the discretion of the investigator in 6.1% of patients, on patient’s request in 3.6%, and due to a loss to follow-up in 1.6%.
During the 12-month study period, 32 patients died. No reason was reported (death NOS) in 6 cases. Thirteen of the remaining 26 patients (50%) died due to tumour progression, and 2 more patients died due to other malignancies. Renal insufficiency was fatal in 1 patient, and 2 patients died from infections. In 5 patients, the cause of death was not specified: 1 patient was hospitalized due to dyspnoea and died on the same day; 1 patient died either of concomitant bronchial carcinoma or pneumonia, as suspected by the investigator who could not provide further details; and general physical health deterioration with fatal outcome was documented in 3 patients. Cardiovascular death was infrequent (n = 3): 1 patient died during mitral valve reconstruction; 1 died due to atrial fibrillation; and 1 died due to circulatory failure. In addition to these 32 patients, the death of 2 patients was reported >30 days after the end of the study period (disease progression and death NOS, respectively). Furthermore, 4 patients excluded from the SAS for different reasons (no drug received, informed consent missing [×2], and good clinical practice violation at the site) died without specified cause, of PCa (×2) or of asthenia.
Discussion
Treatment decisions must be based upon the best levels of evidence. When translating the evidence and the resulting recommendations into an individual treatment decision, it must be remembered that patients included in phase III trials represent a highly selected cohort, regarding age, performance status, pre-treatment situation, and awareness of being part of a scientific experiment, which every interventional study is. In conclusion, it is highly debatable if these results can be directly translated to real-world patients being treated under much less controlled circumstances.
Despite their limitations due to the non-interventional design and limited financial resources, observational studies broaden our existing knowledge, in particular on rare side effects, or address further important questions and aspects related to a given therapy not adequately covered by existing phase III trials. The LEAN study not only addressed basic information on efficacy and toxicity but also provided in-depth information from physicians and patients concerning comorbidity and lifestyle issues. One focus of the LEAN study was to assess treatment indication in relation to current guideline recommendations. Overall, >95% of indications in this study were in line with recommendations for ADT use in the German S3 guidelines, indicating a high level of guideline adherence, also in routine clinical practice (Table 2) [4].
Patients included in this observational study tended to be approximately 1 decade older and had higher comorbidity compared with patients included in contemporary phase III trials in hormone-naïve PCa [10-13]. This difference is also reflected by the distribution of ECOG status [13]. Within this context, the consideration of treatment toxicity is especially relevant in this population. On the other hand, the LEAN cohort included fewer high-risk tumours according to high Gleason score, PSA level, and tumour stage, which would be relevant looking at tumour progression rates [10, 11].
As most information on ADT in PCa patients is derived from studies in patients with metastatic disease, it is important to note that the group of patients with locally advanced and metastatic disease (T4 and/or N+ and/or M+) in this contemporary cohort comprises less than one-third of all patients and approximately one-half if T3 disease is also considered (Table 2). In consequence, the number of patients receiving ADT as an (neo)adjuvant measure or – much more frequently – palliation for biochemical PSA relapse after RP or radiation therapy has dramatically increased and now represents an equally frequent indication for ADT (Table 2). These changes are reflected by the fact that baseline PSA levels were lower in the LEAN study as opposed to phase III studies in metastatic patients [10-13].
Further indications comprise patients with high-risk PCa not suited for definitive therapy. Only 3.9% of patients with relevant data available did not have an obvious clinical indication for ADT. This finding does not suggest malpractice, as relevant information may be missing. Moreover, no data were collected on patient preference in treatment selection, which may play a significant role in selecting ADT as a less invasive treatment in localized disease.
In the overall patient population, the median PSA levels of ≤0.3 ng/mL were achieved after 6 months of treatment and were sustained below this level for the duration of the study (Fig. 3). These observations support the conclusion that ADT works well in a real-world scenario.
Median testosterone serum levels decreased to <0.2 ng/mL 1 month after administration of the leuprorelin implant, with around two-thirds of patients maintaining testosterone below this cutoff for the study duration (12 months) based upon analyses conducted by local laboratories. Applying a testosterone cutoff of 0.5 ng/mL, 92–99% of patients had testosterone levels below the cutoff at different time points during the study, which closely matches results from previous studies [6, 14-17]. The testosterone decrease in LEAN is comparable with the findings in the PR-7 trial (Table 4) and reflect the effectiveness of LHRH agonists in a real-world setting [18]. Another study (ICELAND) found fewer breakthroughs compared with LEAN and PR-7; however, in this study, all patients were treated with leuprorelin for 6 months and bicalutamide for 1 month before study start [19, 20]. In LEAN, only 26% of patients were on ADT within 6 months before study start.
In recent years, the optimum testosterone level for ADT has become controversial. While the castrate level of testosterone has previously been defined as <0.5 ng/mL (1.7 nmol/L), recent studies reported a correlation between lower testosterone levels and overall survival [18]. Based upon these findings, the European Association of Urology (EAU) revised the testosterone target level to <0.2 ng/mL [3]. This contrasts with the current German S3 guideline suggesting an interval not exceeding 0.2–0.5 ng/mL [4], while the National Comprehensive Cancer Network (NCCN) still maintains a target level of <0.5 ng/mL.
There are 3 reasons underlying these discrepant recommendations: (1) in phase III trials, centralized examination has been performed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) as a standard method to measure testosterone due to its superior accuracy, especially at low levels [16, 21]. However, in daily routine, chemiluminescent assays (CLIAs) are used in clinical laboratories worldwide, because they are automatable, fast, sensitive, and inexpensive. A recent study by Morote et al. [17] suggests that testosterone determination using CLIAs yields values that are far higher than those obtained with standard LC-MS/MS. Based on these findings, testosterone measurements obtained in clinical routine may overestimate the true values as determined by LC-MS/MS and thus underestimate ADT effect. (2) The time point of determining testosterone levels has never been defined and is rarely reported. In contrast to the assumption that testosterone levels are taken at the time of the next administration, some studies report a different procedure. For example, in one study, testosterone levels were measured 8–12 weeks after the 3-month LHRH agonist [22]. Observations made in LEAN suggest that testosterone levels might be lower at this time point than at re-administration of ADT. (3) Current evidence suggests that there is no obvious cutoff but rather a linear correlation between the efficacy of testosterone depletion and development of castration-resistant PCa [6, 17, 18, 23].
While SAE information is clearly defined and appears to work reliably in non-interventional studies, reporting of AEs remains a particular limitation of this type of study for several reasons: (1) patients not specifically asked and examined for a specific side effect may not mention it as long as it will not limit them in their daily life; (2) patients receiving routine treatment may not self-inspect themselves to the extent they might when being part of a scientific experiment; (3) some side effects may be so common that they are only documented by the doctor when serious; and (4) the time-consuming reporting of an AE may provide another obstacle, preventing comprehensive documentation, especially of non-serious and lower-grade side effects according to the Common Terminology Criteria for Adverse Events (CTCAE) classification [24]. Hot flushes and hyperhidrosis may serve as an example for these limitations: only 43 and 12 AE reports were received for these well-known side effects, respectively, which clearly contrasts with reports from previous phase III trials and clinical experience that most patients receiving ADT suffer from grade 2 hyperhidrosis. It may be the case that hot flushes/hyperhidrosis are accepted by most patients and urologists as an integral part of therapy and only grade 3 AEs will be recorded. Therefore, less serious AEs may be underestimated in observational studies.
Another way to estimate therapy-related toxicity in an observational study is patient-based premature termination of therapy. This decision must be taken as a clear expression of the fact that patient life is affected by the therapy in an unacceptable way. The observation that only 3.6% of patients chose to terminate ADT early suggests good tolerability of the leuprorelin implant.
Observational studies are known to have relevant limitations related to the non-interventional design and the fact that they do not compare different treatment strategies head-to-head. Furthermore, financial resources are limited and several features needed for high-quality clinical studies are lacking, such as pre-defined study visits, central laboratory, or review of pathology/radiology data. In consequence, definitive answers to open clinical questions must be sought in scientific experimentation in phase III studies or even meta-analyses. However, results from this observational study support the need to validate information from phase III studies, as we have demonstrated that this contemporary middle European patient cohort clearly differs from those patients included in current phase III trials. Finally, much information, such as on treatment indications and guideline acceptance in clinical practice, can only be obtained from observational studies.
Conclusions
This analysis describes a representative Central European PCa patient population receiving leuprorelin solid implant in a real-world scenario. The study demonstrated that the use of ADT in routine clinical practice has clearly changed to earlier tumour stages but closely follows current guidelines. Overall, this interim analysis confirmed previous efficacy findings from phase III trials in a real-world scenario, and no new safety concerns were identified [25]. Meanwhile, the LEAN protocol was amended to allow the inclusion of patients who received ADT in combination with chemotherapy; this extension study is currently ongoing.
Acknowledgements
The authors would like to thank participating patients and their families and participating centres and investigators as well as the responsible staff/study nurses. Editorial support was provided by Olga Ucar of Spirit Medical Communications Group Ltd., funded by HEXAL AG/Sandoz.
Statement of Ethics
All patients have provided written informed consent. The study protocol has been approved by the ethics committee of the Scientific Medical Lead (BSD), that is, the Ethics Committee of the Bavarian Chamber of Physicians, Munich, Germany, and subsequently by the ethics committees of all study sites.
Conflict of Interest Statement
B.J.S.D. has acted as a consultant or assessor for, and has received fees from, Hexal, Janssen, Astellas, and AstraZeneca; S.M. has acted as a consultant or assessor for, and has received fees from, Hexal; C.L. has acted as a consultant or assessor for, and has received fees from, Janssen, Sanofi, MSD, Roche, Astellas, Bristol, and Eusa; N.B.S. has no potential conflicts of interest to declare; E.B. has acted as a consultant or assessor for, and has received fees from, Janssen and Hexal; R.S. is an employee of Sandoz; B.O. was an advisor to Hexal. O.W.H. declared no conflicts of interest.
Funding Sources
This study was funded by HEXAL AG/Sandoz.
Author Contributions
B.J.S.D.: planning and management of study, data analysis, and manuscript drafting. S.M., C.L., N.B.S., and R.S.: data analysis and manuscript drafting. E.B.: manuscript drafting. B.O.: participated in planning, managing, and analyzing the study and in the draft and review of the manuscript. O.W.H.: participated in drafting and reviewing the manuscript.